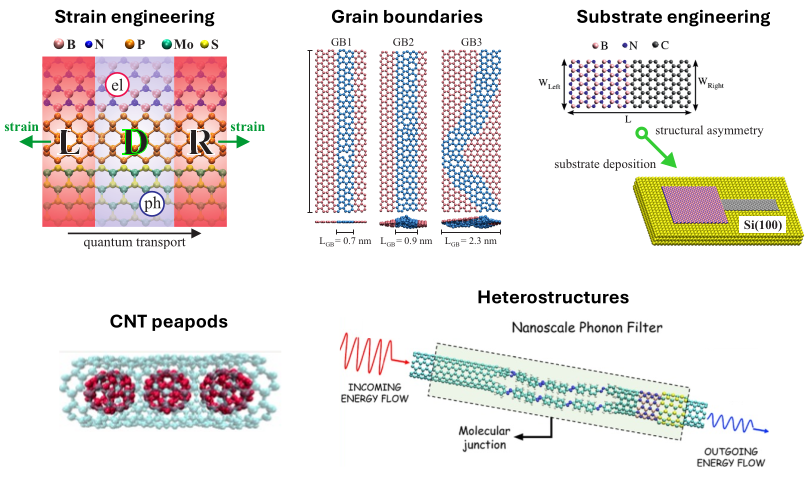
EquiDTB: Augmenting DFTB with machine learning
Density functional tight-binding (DFTB) method is a semi-empirical method that provides access to electronic, structural, and vibrational properties of large and complex molecules requiring only a fraction of the computational cost of density functional theory methods. EquiDTB model has improved the accuracy of DFTB3 method, achieving hybrid DFT-PBE0 level, by replacing the standard pairwise repulsive potentials with many-body equivariant neural network potentials. I have created the EquiDTB Github repository where you can find the NN potentials and scripts to run simulations of molecular systems.
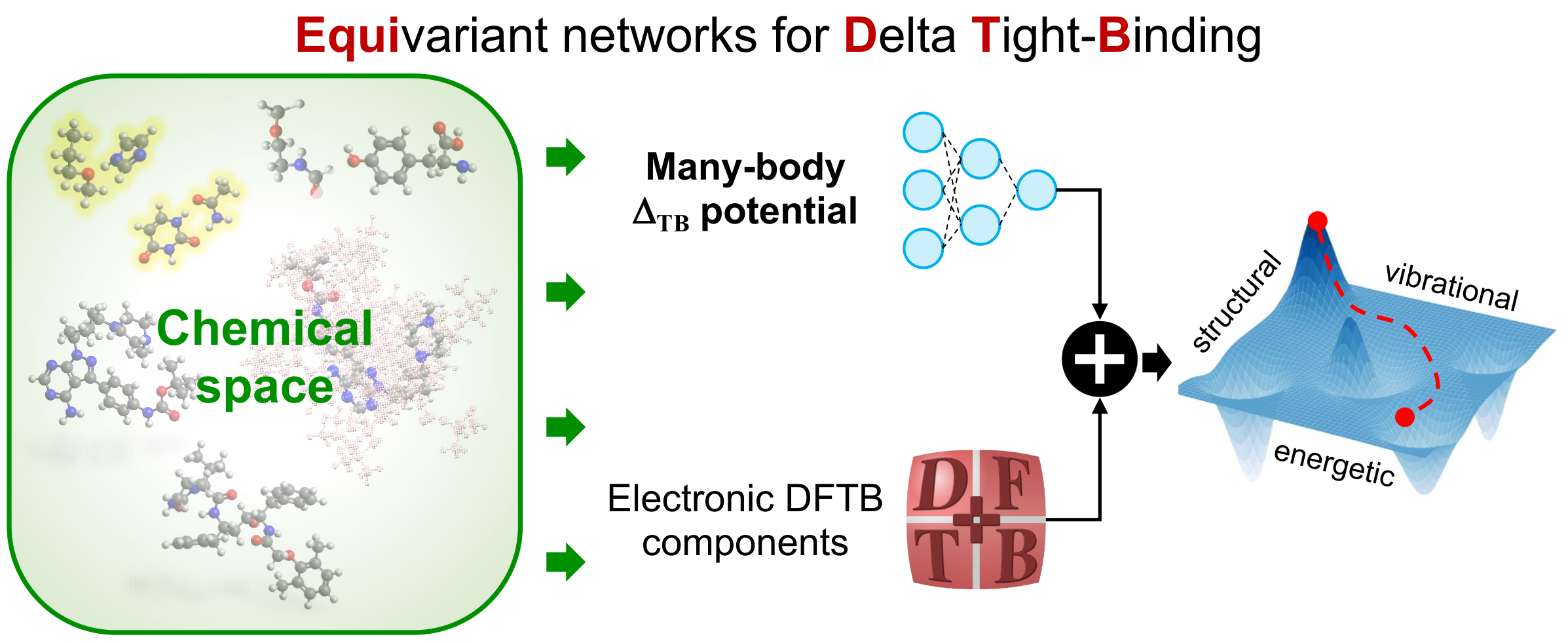
Quantum-mechanical datasets
Implementing robust pipelines for generating comprehensive and highly accurate QM datasets of drug-like molecules/biomolecules is of crucial relevance for the development of reliable AI models. During the last years, I contributed to the generation, curation, formatting, and analysis of extensive benchmark datasets of organic molecules such as QM7-X, Aquamarine, GEMS, QUID, and MORE-Q. More details about these datasets can be found in the associated publications.
“Freedom of design” in chemical spaces
Comprehensive analyses of large datasets are crucial for determining structure-property and property-property relationships of organic molecules. Recently, I exhaustively examined QM7-X dataset, finding an intrinsic “freedom of design” in the molecular property space spanned by small organic molecules, and uncovering molecular design rules that follow physical and chemical intuition. These insights were later used to the successful implementation of a generative framework using variational autoencoders (known as the QIM model), which allowed to map QM properties to 3D structures for small molecules.
ADMET prediction
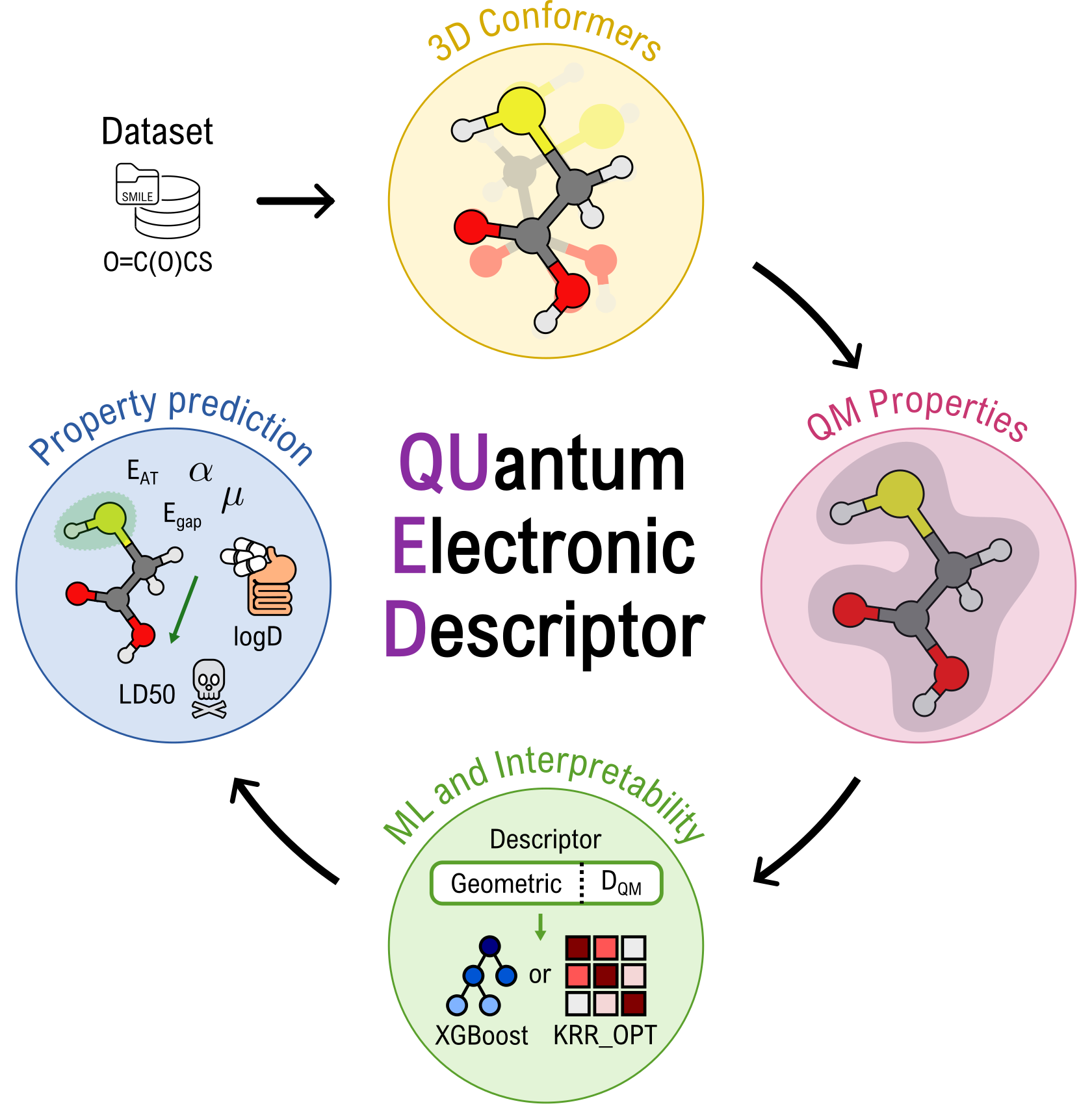
A central challenge in computer-aided drug discovery is the identification of molecular descriptors that can effectively capture both geometric- and electronic structure-derived features, enabling the development of reliable and interpretable predictive models. While numerous descriptors focusing solely on structural characteristics have been recently proposed, improvements in model accuracy often come at the cost of increased computational demands, thereby restricting their practical applicability. To address this challenge, we have developed the “QUantum Electronic Descriptor” (QUED) framework, which integrates both structural and electronic data of molecules to develop ML regression models for property prediction (see QUED Github repository).
Transport in low-dimensional materials
A key step toward improving thermal energy harvesting is the development of atomistic design strategies for smart nanodevices and nanomaterials. Achieving this will require combining computationally efficient atomistic methods with both quantum- and classical-transport approaches. In this regard, we have employed nonequilibrium molecular dynamics (NEMD) simulations to investigate the influence of heat flux asymmetries in thermodynamic properties. Moreover, to address quantum ballistic thermal transport in nanoscale systems, we implemented a nonequillibrium Green’s functions (NEGF) treatment of transport combined with a density-functional tight-binding method (known as the PHONON tool in the DFTB+ code).